




LA DREPANOCYTOSE par le Dr Guy ALOVOR
Le sujet relève d’une importance capitale de santé publique pour le monde noir et l’humanité en général puisqu’il s’agit de la première pathologie génétique en nombre de sujets atteints que ce soit en Afrique, en France, en Europe ou à l’échelle mondiale.
PLAN
1. Prolégomènes
a. Terminologie négro-africaine
b. Importance du sujet
c. « Race » et drépanocytose
d. De l’étymologie et du jargon médical
e. Répartition à travers la planète
2. Historique
a. Imposture habituelle
b. Première maladie génétique dans l’histoire de l’humanité
3. Biochimie et biologie moléculaire
a. C’est quoi l’hémoglobine ?
b. Sa synthèse dans l’organisme
c. La cause de l’hémoglobine S
4. Le diagnostic prénatal
a. Les possibilités de la médecine actuelle
b. Les probabilités de survenue dans la descendance
5. Les fameux « conseils génétiques »
6. Les manifestations cliniques chez les sujets SS
a. Physiopathologie
b. Clinique proprement dite et diagnostic formel
c. Le sport et la mutation drépanémique
7. Le diagnostic
a. Microscopie standard (frottis sanguin)
b. Electrophorèse de l’hémoglobine
8. Le traitement
a. La phytothérapie
b. La spagirie
c. La médecine occidentale
d. Les voies de recherche
9. le cas particulier de l’appareil locomoteur (manifestations ostéo-articulaires)
10. Conclusion.
1. Prolégomènes
- a. Terminologie négro-africaine
Avant de laisser chacun dire le nom de cette mutation génétique qui a toujours existé en Afrique avant l’intrusion des envahisseurs qui ont comme à l’accoutumée attribué la paternité de sa description à un des leurs au début du 20ème siècle, il nous sied de dire que les eυe (éwé) du Golfe de Guinée par exemple la nomment nuɖui. Ce terme traduit le caractère rhumatismal des manifestations périphériques ostéo articulaires survenant par crises dites vaso occlusives. Les Africains en avaient même établi le caractère génétique par la reconnaissance de familles et de lignées drépanocytaires. Certains peuples comme les Yoruba (Nago) ou les Igbo du Nigéria actuel rendent un culte au sujet SS disparu. En effet, les sujets SS qui développent la « maladie » ont une espérance de vie courte mais dépassant la puberté et donc constituent une sorte de « gardiens de la perpétuation du gène muté » en assurant une descendance AS ou SS nécessairement. Or c’est ce qui assure la fameuse protection contre le parasite du paludisme endémique de l’Afrique intertropicale et ailleurs depuis le début de l’humanité. Nous supposons que chaque Africain connaît quelqu’un dans son entourage qui porte des scarifications à la partie gauche du ventre pour guérir une « masse abdominale » : c’est la splénomégalie (augmentation de volume de la rate) chronique des sujets touchés due à une séquestration permanente des globules rouges déformés causant une anémie (diminution de la concentration d’hémoglobine dans le sang). Nous nous souvenons encore de certains de nos camarades de jeu d’enfance en Afrique qui redoutaient les orages au cours desquels la baisse de la pression d’oxygène dans l’atmosphère déclenche les crises douloureuses. Nous y reviendrons plus loin dans la clinique.
NB : Les termes comme « émassi » en mina / guen au Togo relèvent d’une forme d’illettrisme en langue africaine car il s’agit d’un emprunt vulgaire au français « hématie » qui veut dire globule rouge tout simplement sans allusion à sa normalité ou non.
- b. Importance du sujet
Le sujet relève d’une importance capitale de santé publique pour le monde noir et l’humanité en général puisqu’il s’agit de la première pathologie génétique en nombre de sujets atteints que ce soit en Afrique, en France, en Europe ou à l’échelle mondiale ! Nous vous épargnons les statistiques que tout le monde peut consulter sur internet.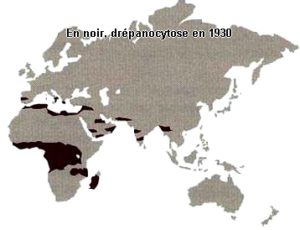
Cependant, il est intéressant de noter que chez les « non Africains » au sens politico-administratif et non anthropologique du terme, la drépanocytose persiste dans le Sud de l’Italie (en Sicile), en Grèce, en Albanie, en Turquie, au Nord d’Israël et dans tout le Moyen-Orient. Cela peut correspondre respectivement aux expéditions des Noirs (Maures ouSarazins), aux Colchidiens issus des troupes du Pharaon Sésostris comme rapporté par Hérodote dans son livre II, aux pré hellènes noirs de l’antiquité, aux survivants du génocide des Cananéens perpétré par Josué et ses hommes lors de la conquête du pays de Canaan, et à la traite transsaharienne de 1000 ans. Nous signalons que la mutation a été retrouvée sur l’ADN d’au moins 3 momies égyptiennes de plus de 3500 ans avant notre ère et il y en a certainement plus si on a le courage de faire pareilles recherches sensibles puisque près de 40% des nègres, naturels d’Afrique comme dirait C. A. Diop, sont porteurs de cette mutation génétique dans les temps modernes où des campagnes d’éradication dupaludisme ont eu lieu. Quant à l’Inde et au Pakistan, c’est chez les plus anciennes populations dravidiennes auteurs de la civilisation brillante de la vallée de l’Indus sur plus de1200 sites (Dholavira, Ganweriwala, Harappa, Lothal, Mohenjo-daro, Rakhigarhi…), aujourd’hui devenus les intouchables et les Tamouls, que l’on retrouve la mutation. Il en est de même au Yémen et en Arabie Saoudite au sein des plus anciennes populations venues d’Abyssinie (Ethiopie).
- c. « Race » et drépanocytose
Cette mutation génétique est le résultat d’une adaptation d’Homo sapiens sapiens remontant à une époque inconnue de sa préhistoire (probablement vers -150 000 à -200 000 ans) lui ayant permis de résister naturellement au parasite Plasmodium falciparum véhiculé par le moustique anophèle femelle et responsable du paludisme. En effet, le portage sain de la drépanocytose, rend les globules rouges difficiles à une infestation par le plasmodium malgré les piqûres de l’anophèle femelle. On estime la protection naturelle à 90% chez le sujet AS. Beaucoup de thérapeutiques n’ont pas une pareille efficacité sur le marché pharmaceutique mondial actuel.
Sur un plan anthropologique, tout individu qui paraît être un naturel d’Europe, d’Asie ou d’ailleurs et chez qui on trouverait un portage sain de la drépanocytose, et il en existe plus qu’on ne le croit, a un ascendant négro africain jusqu’à preuve du contraire. Ceci constitue un des rares marqueurs « raciaux » assez spécifiques mais non sensibles selon le docteur africain américain Charles Finch dans son excellent ouvrage que nous recommandons: « The african background to medical science, essays on african history, science and civilizations = L’origine africaine du savoir médical, essais sur l’histoire, la science et les civilisations africaines ». Nous précisons bien qu’il n’existe pas de marqueur biologique assez sensible de race car même au sein des mélanoïdes, tous ne portent pas la mutation drépanocytaire au point où rien ne permet de deviner à partir de la biologie hormis la drépanocytose, le phénotype (aspect physique) d’un individu. La « race n’existe pas » conclura Diop dans son érudition. Par le simple fait qu’on peut retrouver cette mutation chez d’autres « races », et même si nous savons que c’est dû à un contact avec un mélanoïde, la race devient inexistante encore une fois. Mais les propos ci-dessus sont corroborés par le fait que cette mutation a été portée par Homo sapiens sapiens mélanoïde seul humain sur terre pendant au moins 170 000 ans avant que l’homme de Cro-Magnon, premier ancêtre du leucoderme n’advînt au monde vers -30 000 ans à l’ère würmienne.
- d. De l’étymologie et du jargon médical
Le jargon médical par lequel la science occidentale actuelle ayant pris son essor seulement après ce qu’il est convenu d’appeler le Moyen Age européen, désigne cette mutation est un grécisme récent lorsque l’on a observé dans le sang des sujets affectés, la configuration caractéristique des globules rouges en forme de banane, de croissant, bref de faucille. En effet, le terme DREPANOCYTOSE est composé de trois lexèmes : delta-ro-epsilon-pi-alpha-nu-[ou –êta] omicron-nu (δρέπανον drepanon ou drepanè = faucille) et kappa-upsilon-tau-omicron-sigma (κύτος = cellule) ; on y adjoint ose signifiant maladie. Ce qui stricto sensu est un peu erroné puisque les globules rouges ne sont pas à proprement parler des cellules à l’état adulte puisque dépourvus de noyau cellulaire sauf leurs embryons qui en ont et qu’on appelle des érythroblastes (jeunes cellules rouges).
Les Anglo-Saxons à qui l’on doit la lettre S (Sickle = faucille) ne font pas mieux avec le terme « sickle cell disease ». Cela a donné la SICKLEMIE en français par ailleurs. Rien ne nous interdit dans ces conditions confuses de proposer DREPANEMIE, un néologisme qui nous paraît plus cohérent dans la mesure où il fait référence à la faux dans le sang et mieux, à une anémie falconée ou falciforme. Nous y reviendrons dans les manifestations biologiques hématologiques. L’honnêteté nous oblige à reconnaître la justesse de sicklémie si tant est que ce terme ne fait pas allusion à des cellules, ce qui n’est en tout état de cause pas le cas. En revanche DREPANOCYTEMIE (cellules en faux dans le sang), un autre néologisme de notre part ne sied pas. A la place HEMODREPANOSE convient bien c’est-à-dire un nosos (maladie) liée à une déformation falciforme des hématies. Il en sera de même pour DREPANOPATHIE, c’est-à-dire une passion, une souffrance liée à ces fameux globules rouges en forme de croissant, de banane… et ainsi de suite. Libre à votre imagination à partir de nos langues négro-africaines.
Pour les besoins de cet article grand public, le terme drépanocytose sera utilisé.
La drépanocytose fait partie de la grande famille de ce qu’on appelle les HEMOGLOBINOPATHIES ou HEMOGLOBINOSES par mutation génique (S, C, D, E…selon parfois le lieu de description « princeps ») ou par insuffisance de fabrication par l’organisme (alpha et bêta thalassémies ainsi dénommées à causse de leur répartition autour du bassin méditerranéen : mare medi terra = mer du milieu de la terre ; si vous avez déjà fait de la thalasso = mer). Le dernier cas constituant une absurdité sémantique dans le sens où thalassémie renvoie à l’idée d’avoir « de la mer dans le sang ». Nous proposons le terme simple d’insuffisance par délétion hémoglobinique alpha ou bêta.
Hémoglobinopathie = hémoglobine et pathos (passion, souffrance).
Hémoglobinose = hémoglobine et nosos (maladie).
- e. Répartition à travers la planète
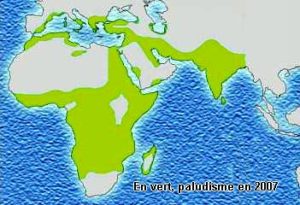
Ci-dessous les répartitions géographiques de la drépanocytose, des thalassémies et du paludisme. En noir répartitions avant 1930, début des campagnes d’éradication du paludisme.
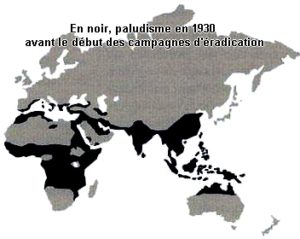
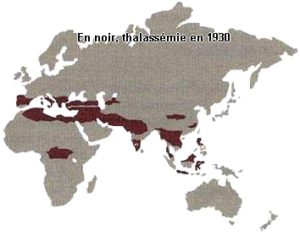
Bien sûr il faut ajouter la présence de la mutation en Amérique du Nord, Centrale, du Sud et dans les Caraïbes qui est due à l’odyssée précolombienne des mélanoïdes et évidemment à la tragique déportation par les Européens des Africains.
2. Historique
- a. Imposture habituelle
Comme à l’accoutumée, les Occidentaux se sont attribué la paternité de la description d’un phénomène naturel extérieur à leur environnement et à leur univers culturel. C’est ainsi que la littérature médicale distille que c’est à Chicago qu’un certain Dr Herrick aurait décrit en 1910 les globules rouges caractéristiques chez un étudiant noir en chirurgie dentaire originaire de la Grenade qui souffrait de douleurs, de fièvre et d’anémie. En fait ce serait son interne Irons qui aurait observé au microscope des globules rouges en forme de faucille et de feuilles d’acanthe. La malhonnêteté intellectuelle doublée d’une avidité de gloire ont conduit à ne pas associer le pauvre interne Irons à la publication du cas. C’est ce qu’en rapporte en tout cas l’histoire.
Ci-dessous image au microscope standard d’un frottis de sang.
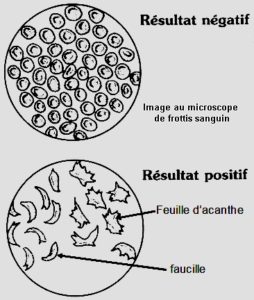
D’autres descriptions ont eu lieu avant, aux 18ème et 19ème siècles chez des populations d’Afrique avec des douleurs, une anémie et des syndromes infectieux.
- b. Première maladie génétique dans l’histoire de l’humanité
Nous passons les différentes péripéties pour signaler que ce ne sera qu’en 1957 qu’avec l’essor de la biologie moléculaire, que l’hypothèse d’une mutation ponctuelle au niveau de l’ADN a été évoquée puis confirmée faisant ainsi de la drépanocytose la première modification protéique humaine (HbS) conséquence d’une mutation au niveau d’un gène alors que les biologistes s’attendaient à faire la description princeps d’une modification génotypique entraînant une modification protéique chez un virus ou au mieux chez une bactérie, organismes beaucoup plus élémentaires, moins complexes. On peut dire que c’est un don de l’homme noir au progrès scientifique de l’humanité à signaler si besoin est, auquel on peut ajouter au passage les cellules immortelles de l’Africaine Américaine Henrietta Lacks disparue depuis 1951 que tous les laboratoires du monde entier s’arrachent pour avoir du matériel humain vivant aux fins de recherches médicales.
3. Biochimie et biologie moléculaire
Faisons connaissance avec l’hémoglobine, son rôle, sa synthèse et l’origine de la drépanocytose.
- a. C’est quoi l’hémoglobine ?
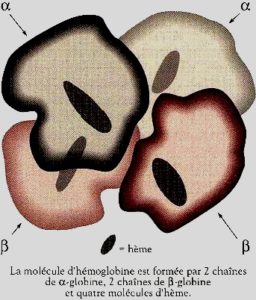
L’hémoglobine est une protéine (chaîne d’acides aminés) complexe pigmentée située sans les globules rouges et responsable de la couleur rouge du sang des vertébrés. Elle est constituée de deux paires de chaînes de globuline (aspect tridimensionnel de globe, les plus grosses protéines du sang par opposition à l’albumine qui évoque étymologiquement le blanc de l’œuf: albus en latin voulant dire blanc d’où albinos…) et de 4 molécules métallo organiques (tout ce qui est à base de carbone, d’hydrogène, d’oxygène et d’azote) appelées hème ayant en leur centre un ion ferreux Fe++ à l’état normal (ou ferrique Fe+++ donnant la méthémoglobine pathologique). Elle est responsable du transport du dioxygène O2 par les globules rouges (hématies) destiné aux tissus des organismes vivants en vue des réactions d’oxydation indispensables à la vie. On parle ainsi du pouvoir oxyphorique (transport de dioxygène) de l’hémoglobine. Ceci correspond à la quantité de dioxygène (O2) qu’elle peut fixer. Cette valeur est d’environ 1,34 ml d’O2/g d’hémoglobine.
On distingue des chaînes de globuline α, β, γ, δ. L’hémoglobine normale de l’Adulte est constituée de 2 chaînes alpha et de 2 chaînes bêta. Elle est symbolisée par HbA1 qui représente 98%, les 2% restants sont constitués d’hémoglobine HbA2 constituée de 2 chaînes alpha et de 2 chaînes delta. Chez le fœtus et le nouveau-né, on a deux chaînes alpha et deux chaînes gamma de globuline, c’est l’hémoglobine fœtale HbF, qui a la propriété d’avoir une plus grande affinité pour le dioxygène.
C’est le lieu de faire un aparté sur l’hémoglobine glycosylée HbA1c, comportant une fixation d’une molécule de glucose et qui est une sorte de « mouchard » biologique de bonne observance ou non du traitement chez le diabétique dans la mesure où ce marqueur permet de révéler si la glycémie (concentration de glucose dans le sang) des derniers jours avant la prise de sang pour le dosage de l’HbA1c a été régulièrement normale. Les malins qui ne suivent pas le régime alimentaire antidiabétique et le traitement et qui s’arrangent pour avoir une bonne glycémie de façade le jour du contrôle auprès du médecin diabétologue ne trompent qu’eux-mêmes.
- b. Sa synthèse dans l’organisme
Synthèse de l’hémoglobine et origine de l’hémoglobine S. L’ADN (DNA en anglais), Acide Désoxyribonucléique est le constituant biochimique du chromosome des êtres vivants. Chez l’homme on compte 23 paires de chromosomes localisées dans le noyau des cellules, chaque paire est le fruit d’un chromosome provenant du père et d’un autre provenant de la mère qui se sont brassés puis redistribués aléatoirement. C’est ce qui explique que chaque individu est différent et unique. Les gènes qui sous-tendent les caractères exprimés sont situés sur ces paires respectives, chaque gène est ainsi constitué d’un assemblage de 2 « hémi gènes » qu’on appelle des allèles. L’ADN est constitué de 2 chaînes moléculaires issues d’une succession de molécules appelées des nucléotides constitués de 4 bases azotées, 2 puriques et 2 pyrimidiques (Adénine, Guanine, Thymine, Cytosine) :
- · le désoxyadénosine mono phosphate (dAMP), l’Adénine (A) étant la base purique le composant ;
- · le désoxyguanosine mono phosphate (dGMP), la Guanine (G) étant la base purique le composant ;
- · le désoxythymidine mono phosphate (dTMP), la Thymine (T) étant la base pyrimidique le composant ;
- · le désoxycytidine mono phosphate (dCMP), la Cytosine (C) étant la base pyrimidique le composant.
Ces nucléotides ont la particularité de s’unir deux à deux par complémentarité au niveau de leur base azotée constitutive:
- · le dAMP (Adénine) avec le dTMP (Thymine) en établissant deux liaisons hydrogène ;
- · le dCMP (Cytosine) avec le dGMP (Guanine) en établissant trois liaisons hydrogène.
NB : L’uridine mono phosphate (UMP) est un nucléotide présent uniquement dans l’ARN (Acide Ribo Nucléique), il remplace le dTMP (Thymine) qui n’apparaît que dans l’ADN. L’UMP s’apparie avec l’adénosine mono phosphate (AMP) dans l’ARN.
Cela donne pour l’ADN des chaînes ayant la forme suivante :
A G T C………T C C G….
II III II III II III III III
T C A G………A G G C…
Il faut savoir que toutes les cellules d’un organisme vivant ont le même génome et peuvent donc potentiellement fabriquer les mêmes protéines, expressions des gènes. Mais alors, comment se fait-il donc que par exemple, les cellules de la glande pinéale ne fabriquent pas les mêmes substances que celles du pancréas ou de la peau ? Cela résulte de la spécialisation cellulaire issue de la différenciation par un jeu de répression et d’ampliation / expression génique. C’est la réversion de ce processus naturel qui est la base des phénomènes de clonage qui shuntent l’étape initiale du brassage chromosomique faisant suite à la fécondation et qui permet la singularisation de chaque individu au sein d’une espèce. Autrement, tous les enfants d’un couple seraient identiques comme des clones. Cette réversion permet d’obtenir à nouveau une cellule indifférenciée totipotente (pouvant évoluer vers tous les tissus) et ainsi de suite. Le seul hic, c’est qu’on a remarqué que le clone obtenu garde en mémoire l’âge de la cellule « mère ». Par exemple la brebis Dolly était née avec de l’arthrose (usure de cartilage) !
Comment se fait la synthèse protéique à partir des gènes ? Un gène est dit dominant lorsque un seul allèle suffit à son expression et récessif lorsqu’il en faut deux. Il peut être autosomique lorsque situé sur une paire chromosomique autre que la 23 ou gonosomique lorsque porté par la 23ème (XX ou XY). Pour ce faire, l’ADN se comporte comme la gamme diatonique issue de la vallée du Nil (attribuée évidemment à Pythagore Nemakos de Samos), avec les 4 nucléotides dont chaque combinaison ordonnée de 3 tels une grille d’accord de guitare donne une note qu’est un acide aminé, élément constitutif des chaînes protéiques. Avec les 4 nucléotides, on fabrique les 20 acides aminés courants provenant des 64 combinaisons possibles (avis aux arithméticiens ; dénombrez la quantité de triplets possibles à partir de 4 nucléotides en sachant que l’ordre importe, chaque nucléotide pouvant être répété; cela revient à un Arrangement avec répétition 3 à 3 avec la formule nk. Ici, cela revient à 43 = 64 possibilités). Chaque triplet de nucléotides s’appelle un codon, terme inventé par le biologiste sud africain Sydney Brenner en 1960 pour la petite histoire.
Traduction des codons en acides aminés dans le code génétique
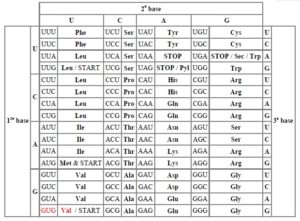
Ala alanine Gln glutamine
Arg arginine His histidine
Asp aspartate ou acide aspartique Ile isoleucine
Asn asparagine Leu leucine
Cys cystéine Lys lysine
Glu glutamate ou acide glutamique Met méthionine
Gly glycine Phe phénylalanine
Pro proline Ser sérine
Trp tryptophane Tyr tyrosine Thr thréonine Val valine
- c. La cause de l’hémoglobine S
La cause moléculaire de la drépanocytose est une mutation ponctuelle (unique !) au niveau du gène à l’origine de la production de la chaîne d’acides aminés formant la bêta globine (ça paraît être une clé simple issue de la Nature). C’est la substitution d’une Adénine par une Thymine en position 20 dans la séquence de nucléotides du gène de la bêta globine qui entraîne une modification de la séquence protéique dans laquelle un Glutamate est remplacé par une Valine en position 6. L’hémoglobine mutante qui en résulte est l’hémoglobine S ou HbS. Ceci se passe au niveau du chromosome 11 sur son bras court. Comme par hasard, c’est le même chromosome 11 qui porte au niveau de son bras long le gène de l’albinisme par défaut de synthèse de mélanine dont la qualité est responsable de la noirceur de la peau. NOUS EN DÉDUISONS ENCORE UNE FOIS LE LIEN NATUREL EXISTANT ENTRE LA PIGMENTATION ÉPIDERMIQUE ET LA DRÉPANOCYTOSE !!!

On compte environ 500 hémoglobines « anormales » !!! par des mutations génétiques ponctuelles (délétions, substitutions ou insertions) dans les différentes variétés de l’espèce humaine.
Au niveau phénotypique, l’usine de fabrication de l’hémoglobine (hémopoïèse plus précisément), est la moelle osseuse. C’est le lieu où des cellules se sont spécialisées, comme nous l’écrivions plus haut, en l’expression protéique du gène de l’Hb.
4. Le diagnostic prénatal
- a. Les possibilités de la médecine actuelle
Diagnostic prénatal Il est possible, lorsque les deux parents sont porteurs de la mutation de proposer un diagnostic anténatal :
- par biopsie de trophoblaste (enveloppe autour de l’œuf embryonnaire) à partir de la11ème semaine de grossesse (13ème semaine d’aménorrhée)
- par amniocentèse (ponction du liquide autour de l’embryon) à partir de la 17èmesemaine de grossesse (19ème semaine d’aménorrhée).
- b. Les probabilités de survenue dans la descendance
Examinons les différents cas de parents et les probabilités des enfants mutants.
![]()
Dans le cas ci-contre, la probabilité de portage de la mutation est de 50%. Si un enfant venait à être SS, il faut se poser des questions sur sa génitrice (des erreurs ont pu se produire dans des maternités ; cf. le film la vie est un long fleuve tranquille d’Etienne Chatiliez) ou son géniteur car sa probabilité théorique est nulle.

Dans le cas ci-contre, tous les enfants à 100% seront porteurs sains AS. Un enfant indemne AA ou drépanocytaire SS posera les mêmes questions de parentalité biologique.

Dans le cas ci-contre, la descendance a une équiprobabilité d’être SS ou AS. Les mêmes doutes seront soulevés pour tout enfant indemne AA.
![]()
Dans ce dernier cas, la descendance a 25% de probabilité d’être indemne, 25% d’être SS et 50% d’être AS. Ce cas est intéressant car il permet de prédire la situation de deux Africains qui font des enfants sans « rien savoir » de leurs antécédents. Puisque nous savons la prévalence du portage sain dans la population africaine qui est de 40%, deux personnes d’ascendance africaine qui font des enfants, ont 70% de probabilité de AA, 20% de AS et seulement 10% de SS. Lorsque le statut d’un ou des deux parents est connu, cela renvoie à un des cas ci-dessus.
Inutile d’ajouter que lorsque les deux parents sont AA ou SS, tous les enfants sont attendus AA ou SS respectivement et ils seront biologiquement curieux autrement.
Cas particulier de parents double hétérozygotes SC: 50% des enfants seront SC, 25% SS et il y aura 25% de fausses couches correspondant au CC qui est létal.
Il est utile de préciser, puisqu’il s’agit d’un phénomène autosomique récessif, que le sexe de la descendance, ni de l’ascendance d’ailleurs, ne compte pas pour toutes les situations évoquées.
5. Les fameux « conseils génétiques »
Nous mettons ceci exprès entre guillemets ces conseils car certaines Africaines et certains Africains de la diaspora notamment porteurs de la mutation se sont entendu conseiller par des médecins de ne pas faire d’enfants avec d’autres Africains au risque de faire des drépanocytaires. Ceci constitue une absurdité phénoménale car si Homo sapiens sapiens avait pris de tels conseils, ces fameux médecins « conseillers génétiques » ne seraient peut-être jamais venus à exister. Même la fameuse OMS qui n’est pas exempte de scandales sanitaires (cf. H1N1), avait eu l’impertinence en 1978 de conseiller aux Africains, une réduction des naissances pour lutter contre l’incidence de la mutation ! C’est scandaleux. D’une part, cette mutation qui remonte à au moins 200 000 ans (âge supposé de l’homme moderne) nous a protégés du paludisme endémique qui tue plus que le VIH actuellement ; d’autre part, même le sujet SS d’une certaine manière en constituant une sorte de réservoir humain de la mutation, participe à la pérennité de l’espèce, justifiant certains cultes comme ceux évoqués dans les prolégomènes puisque durant sa courte existence terrestre, il lui est offert quand même de transmettre la mutation avant de rejoindre son Ka divin. En homme de sapience et de progrès, nous nous posons tout de même la question de la pertinence de l’éradication d’une telle mutation, auquel cas doit-elle aller de pair avec celle du Plasmodium falciparum ? Mais alors cela ne pose-t-il pas des questions plutôt métaphysiques concernant la faune? Le débat est ouvert ce d’autant que pour une mutation ponctuelle comme c’est le cas, la thérapie génique doit paraître simple.
Par ailleurs, comme souligné plus haut, les mariages « inter raciaux » ne mettent pas à l’abri de la mutation drépanocytaire à 100%. Et puis si vraiment on le souhaite, plutôt que de « briser des amours » et de faire une fixation sur un mariage inter racial forcené, il existe toujours en dernier ressort l’interruption thérapeutique de grossesse qui n’a pas de limite légale de délai, le diagnostic prénatal étant possible comme signalé ci-dessus.
6. Les manifestations cliniques chez les sujets SS
Puisqu’il s’agit d’une mutation récessive, seules les formes homozygotes (mêmes allèles mutés S et S ou SS) ou doubles hétérozygotes encore dites hétérozygotes composites (deux allèles mutés différents : SC, SE, SD…Sβthalassémie), présenteront des manifestations cliniques fonctionnelles comme des douleurs ou physiques visibles comme l’anémie hémolytique, la grosse rate, les troubles staturo-pondéraux…
Il serait fastidieux d’énumérer dans le cadre d’un tel propos l’ensemble des manifestations cliniques et para cliniques car il s’agit plus que d’une maladie de système pouvant toucher tous les organes du corps humain, tous les appareils et systèmes du corps physique et métaphysique: neurologique, digestif, respiratoire, musculo-squelettique, génito-urinaire, cardio-vasculaire, immunologique, organes des sens…psychologique. Et même social.
Nous nous permettons de nous appesantir légèrement sur l’appareil locomoteur qui est notre spécialité quotidienne avec les progrès techniques dont peuvent bénéficier les sujets ayant des destructions ostéo-articulaires. Mais avant cela, nous citerons quelques manifestations plutôt singulières puisque tout bon Africain connaît les principales manifestations et que nous ne sommes pas ici pour faire un cours de séméiologie médicale (étude des symptômes).
- a. Physiopathologie
La physiopathologie (dysfonctionnement) à l’origine des troubles observables en pratique médicale procède de la falciformation des globules rouges lorsque la quantité de dioxygène diminue dans le sang (hypoxémie). Ceci peut du reste être mis en évidence in vitro (en dehors de l’organisme). La conséquence est une tendance à l’agglutination des globules rouges falconés et à l’obstruction des capillaires (petits vaisseaux microscopiques distributeurs de l’oxygène aux tissus), d’où les douleurs dites vaso occlusives en cas de crise aiguë et les nécroses (mort tissulaire) à la longue et l’anémie hémolytique (destruction des globules rouges déformés par la rate). Du coup la rate augmente de volume à force de séquestration des globules rouges déformés, c’est la splénomégalie (grosse rate). Le défaut d’oxygénation tissulaire entretien ainsi un cercle devenant vicieux exposant le sujet aux infections.
Mécanismes et enchaînements causals conduisant à l’apparition des troubles: 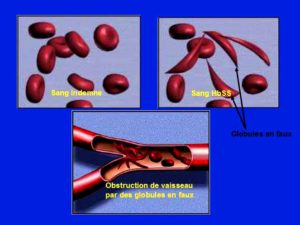
Les manifestations procèdent d’un primum movens conséquence de la propension de l’HbS à polymériser (agglutination par réaction biochimique de monomères, c’est-à-dire la molécule d’HbS) lorsque le dioxygène O2 vient à manquer. Il en résulte une sorte de ratatinement tridimensionnel des globules rouges qui passent d’une forme grossièrement sphérique à une forme de banane ou de croissant de viennoiserie. Les globules rouges ainsi déformés ont eux-mêmes tendance à s’agglutiner dans les petits vaisseaux conduisant ainsi à des micro emboles (obstruction de la lumière des vaisseaux). Inutile d’ajouter qu’il en résulte un cercle vicieux d’aval et ainsi de suite. Lorsque cette chaîne causale est minime, cela reste peu parlant mais participe d’une souffrance tissulaire à bas bruit comme chez les tabagiques ou les personnes qui ont comme on dit du cholestérol. Les tissus présentent des zones de nécrose (mort) qui vont s’additionner à chaque fois. Cela rend les tissus vulnérables vis-à-vis des infections. En cas de démultiplication de la chaîne, on aboutit à la crise douloureuse que nous faisons l’économie de décrire ici. Tout se passe comme ce que l’on observe dans l’angine de poitrine (angor) ou l’infarctus du myocarde (en tout cas sur cœur non transplanté) ou dans la claudication intermittente des membres inférieurs chez les personnes qui ont les « artères bouchées ». Par ailleurs, lorsque le foie et la rate voient circuler des globules rouges ainsi méconnaissables, ils les séquestrent et les détruisent. Cela occasionne une anémie dite hémolytique (destruction des globules rouges) avec un ictère (ou jaunisse) dû à la pigmentation du blanc de l’œil et des paumes et plantes des membres par le produit de dégradation de l’hémoglobine ainsi libérée qu’est la bilirubine produite par le foie en temps normal. La jaunisse est difficile à observer sur peau noire pour les novices. A force de séquestration, le foie et la rate deviennent gros, c’est respectivement ce qu’on appelle hépatomégalie et splénomégalie.
- b. Clinique proprement dite et diagnostic formel
Parmi les manifestations un peu particulières, nous allons citer des cas d’AVC (accident vasculaire cérébral) chez des patients d’âge jeune, des pertes de vue dues à une rétinopathie ischémique (rétine privée de vascularisation), des insuffisances respiratoire, hépatique, rénale et cardiaque par nécroses répétées (infarcissements), le priapisme : du dieu ithyphallique grec Priape réplique du dieu kamite Min qui lui est circoncis (érection douloureuse permanente sans excitation érotique) pouvant aboutir à une souffrance du corps caverneux et à l’impuissance compte tenu de la pudeur à en parler chez le sujet, les problèmes psychiques liés au vécu de la situation, à la relation avec les parents qui considèrent l’enfant avec l’affect de sa faible espérance de vie, aux complications multiples perturbant la scolarité chez l’enfant pouvant occasionner une sorte de nanisme social…
IL EST IMPORTANT DE PRECISER QUE LE DIAGNOSTIC FORMEL ET PRÉCIS EST BIOLOGIQUE ET SE FAIT A PARTIR D’UNE PRISE DE SANG EN DEMANDANT UNE ELECTROPHORESE DE L’HEMOGLOBINE ET NON DES PROTEINES. La confusion qui est souvent faite nous oblige à cette précision.
- c. Le sport et la mutation drépanémique
Il semble que les sujets hétérozygotes AS aient des prédispositions pour de meilleures performances lors d’efforts brefs et explosifs (le sprint en athlétisme par exemple), impliquant le métabolisme anaérobie alactique (sans production de lactate) auquel ces sujets sont habitués. En revanche, les formes homozygotes sont défavorisantes et constituent un risque non négligeable pour ne pas dire vital pour les activités physiques et sportives pouvant déclencher des crises voire des morts subites. Nous conseillons un dépistage par une électrophorèse systématique de l’Hb à tout Africain qui pense au sport de haut niveau. La récente histoire de Lassana Diarra lors du stage en altitude dit d’oxygénation (au fait l’objectif est de profiter de la baisse du taux ambiant de dioxygène pour que les organismes secrètent naturellement une augmentation de globules rouges comme le fait le dopage à l’érythropoïétine : EPO artificielle) de l’équipe de France de foot est riche d’enseignements à ce sujet. Nous ne savons pas si la mort subite en direct à la télé du regretté footballeur camerounais Vivien Foe avait fait l’objet lors de l’autopsie d’une électrophorèse de l’Hb. La famille peut toujours réaliser une recherche généalogique (ascendance et descendance).
7. Le diagnostic
- a. Microscopie standard (frottis sanguin)
La microscopie simple permet sur une goutte de sang prélevé surtout en cas de crise d’observer des globules rouges à l’aspect falciforme. Les éléments d’orientations sont constitués par les antécédents familiaux et l’origine ethnique sur laquelle nous ne revenons pas.
- b. Electrophorèse de l’hémoglobine
L’électrophorèse est une séparation des différentes formes d’une substance par un champ électrique en utilisant leurs variations de propriétés migratoires dans ce champ. Cet examen de laboratoire biologique permet d’isoler les différentes formes d’hémoglobines. La biologie moléculaire (étude de l’ADN) est une débauche biotechnologique inutile dans le cadre de la pratique clinique courante. Elle ne se justifiera que dans le cadre de la recherche médicale et autres manipulations justifiées.
8. Le traitement
a. La phytothérapie
A tout seigneur tout honneur. Des traitements à base de plantes africaines ont été développés de longue date pour palier aux souffrances : Zanthoxylum fagara et ses extraits : c’est un arbuste épineux qui entre dans la composition de plusieurs remèdes.
Eugenia caryophllum
Sorgul bicolor
Piper guineense
Pterocarpus osun
Pois d’Angola : Cajanus cajan
Carica papaya
Alchornea cordifolia
Nous citons deux préparations présentées sous forme galénique industrielle, l’une au Bénin par le Docteur MEDEGAN, le VK500 à base du fameux Zanthoxylum fagara et l’autre au Burkina Faso, dont le promoteur est le Pr. Pierre GUISSOU qui a mis au point un mélange appelé FACA à base de Zanthoxylum fagara et de Calotropis procera. Inutile de vous rapporter le nombre d’articles de détracteurs occidentaux experts de tout et de rien pendant qu’aucune étude africaine ne se livre jamais à ce genre de grandes manœuvres au sujet des diverses substances chimiques douteuses qui sont déversées sur le continent à commencer par les laits artificiels pour bébé causant des décès. Nous vous épargnons ici les corruptions des « grands professeurs » de pharmacologie qui sont au sein des agences de « sécurité » que ce soit l’AFSSAPS en France ou le FDA aux USA avec les scandales gigantesques pendant qu’on donne des leçons de vertu aux « sous-développés » qui ne se livrent qu’à de petits larcins à côté de leurs maîtres dans l’art. Actuellement le contribuable français devra payer plus de 30 000 ré opérations de prothèse mammaires en silicone frelaté pendant que les scandales des laboratoires Servier et leur coupe-faim sont encore pendants. Et nous ne sommes qu’au début. Et comme le fabricant des fameuses prothèses PIP a eu son autorisation de mise sur le marché (AMM) en graissant la patte à qui vous devinez, ni vu ni connu. Sauf que depuis, des bonnes femmes qui ont voulu se faire belles (ce n’est pas un délit), ont vu leurs implants exploser (nous ne savons pas si leurs partenaires n’ont pas été assez doux, fallait les prévenir peut-être) avec des granules de silicone disséminés partout dans le corps et au moins 4 cas de cancer sont à déplorer avec déjà 2 décès à la clé officiellement. Voilà le genre de sécurité sanitaire que nous payons.
- c. La spagirie
D’autres traitements issus de plantes, de minéraux, de métaux, d’animaux sont basés sur ce que les eυe par exemple appellent AMAWƆWƆ et que les mystiques alchimistes gnostiques du moyen âge européen ont appelé depuis les travaux du germanique Paracelse la SPAGIRIE. Les férus de science qui croient systématiser ainsi le corps humain parlent de choses qu’ils ignorent. Mais en fait, pour l’observateur que nous sommes, cet AMAWƆWƆ qui fait appel au caché inconnaissable AMA ou AMON en utilisant les éléments précités et tout un rituel philosophico astronomique est du niveau de la physico chimie quantique tout simplement. Les praticiens eux-mêmes disent se servir de l’énergie vitale contenue dans les éléments pour rétablir l’équilibre à partir du désordre à l’origine des souffrances. Les détracteurs des praticiens africains ne nous démentiront pas qu’ils font en cachette des expériences de mécanique quantique pour percer certains mystères de ce qu’ils appellent les phénomènes paranormaux. Nous vous conseillons l’excellent article de DIOP sur les « crises majeures de la raison » à l’occasion d’un colloque sur la science et la religion à Dakar.
- d. La médecine occidentale
L’hydroxyurée est réputée apporter un bénéfice mais ses effets tératogènes (malformations fœtales) sont rédhibitoires et nécessitent une contraception efficace chez les jeunes filles traitées et une cryoconservation d’ovocytes. Les antalgique majeurs comme la morphine durant les crises douloureuses vaso-occlusives (obstruction des vaisseaux). La greffe de moelle est réputée modifier le phénotype SS et consiste en une destruction chimique de la moelle osseuse puis recolonisation par un greffon allogène. Il existe un risque mortel non négligeable pour ce traitement lourd et coûteux.
Les transfusions sanguines. Le traitement martial (Fer). Pour corriger l’anémie si non bien supportée.
La chirurgie d’ablation de la rate (splénectomie) pour limiter la séquestration des globules rouges et l’anémie.
Le traitement des infections, des différentes insuffisances viscérales, les règles hygiéno-diététiques évitant les sur complications comme les infections, le sport chez le SS ou SC étant à éviter, une alimentation riche en fer (viande rouge surtout de cheval, vin blanc) et l’évitement de l’hypoxie déclencheur des crises relèvent du bon sens.
- e. Les voies de recherche
La thérapie génique pour corriger la substitution nucléotidique à l’origine de l’HbS. En 2001 est publié dans la revue Nature puis Science et Avenir un essai sur 20 souris avec succès dans le cas de la drépanocytose. Des essais sur des singes seraient en cours. En 2007, l’AFSSAPS (Agence Française de Sécurité Sanitaire et des Produits de Santé) a autorisé l’usage du VIH inactivé comme vecteur de grandes quantités de fragments géniques correcteurs dans la moelle osseuse sur un sujet atteint de bêta thalassémie. Il a été guéri selon les résultats.
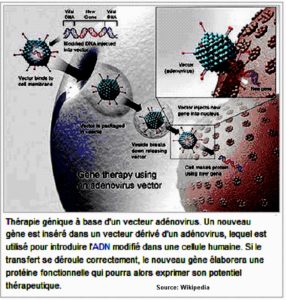
Source : Wikipedia
9. Le cas particulier de l’appareil locomoteur (manifestations ostéo-articulaires)
Nous en arrivons au cas particulier des manifestations ostéo articulaires. Ces complications sont essentiellement les nécroses (mort) osseuses faisant le lit des infections comme l’ostéomyélite (inflammation de l’os et de sa moelle) infectieuse et des destructions articulaires. Pour ces destructions articulaires invalidantes responsables par exemple des boiteries que l’on observe souvent, la chirurgie prothétique a fait de grands progrès ces dernières années. Des biomatériaux de mieux en mieux tolérés (titane, alliages de chrome-cobalt…) associés aux couples de friction articulaire (tribologie) comme la céramique d’alumine et du polyéthylène de haute densité de mieux en mieux dessinés par informatisation et de mieux en mieux usinés donnent une très grande longévité aux implants prothétiques pouvant dépasser 30 ans. Malgré ces progrès, un taux incompressible de 2% d’infections liées aux soins durant la première année (nosocomiale de nosos = maladie et komein = soigner) est à déplorer malgré toutes les précautions d’asepsie et d’antibioprophylaxie. Les équipes ayant un taux plus élevé doivent faire des progrès ! Ce taux doit être certainement plus élevé si l’on considère une population exclusivement constituée de drépanocytaires à cause du terrain favorisant les infections. A la longue, un risque de greffe bactérienne existe lors d’infections d’autres sites (pulmonaires, cutanées, urinaires) entraînant une bactériémie (circulation de microbes dans le sang). Cela peut malheureusement nécessiter une ré opération avec retrait de la prothèse infectée et un changement parfois en deux temps espacés de quelques semaines d’antibiothérapie soutenue et ciblée. Les luxations sont de plus en plus rares au niveau de la hanche grâce aux prothèses à grand diamètre plus proches de l’anatomie normale et aux doubles mobilités permettant une vie active normale et une sexualité sans contraintes positionnelles liées à la prothèse de hanche.
10. Conclusion.
Cette mutation particulière a été un facteur protecteur d’Homo sapiens sapiens mélanoïde dans cet environnement où il est apparu et où existait probablement avant lui le parasite Plasmodium falciparum responsable du paludisme endémique. Tout comme la pigmentation riche en mélanine l’a protégé des UV du soleil selon la loi de Gloher, la Nature qui a créé l’homme moderne l’a doté de cette protection qu’est l’hémoglobine S lui ayant permis une survie avant la mise au point de traitements antipaludéens par lui-même par la suite. Ces deux aptitudes qualités sont portées par le même chromatosome 11 (bras court pour le gène de production de bêta globuline et bras long pour le gène de production de l’enzyme décisive de la mélanine, la thyrosinase). La variété albinoïde apparue après 20 000 ans de séjours dans le froid würmien en -30 000 ans n’ayant plus besoin de ces aptitudes (peau noire et HbS), les a simplement réprimées génétiquement. En conséquence, autant la peau noire n’est pas une tare chromosomique, l’HbS n’est pas une aberration génétique mais bien au contraire, c’est une chance pour le genre humain. Sans ces éléments, l’homme n’aurait pas connu cette belle odyssée planétaire depuis la région des grands lacs africains ayant conduit à l’apparition des diverses variétés de l’Humanité moderne. N’empêche, la souffrance est intolérable et doit nécessiter soulagement par humanisme et ce dans le respect de l’harmonie cosmique maâtique. C’est cela la véritable sapience, la haute science des sages. C’est vrai, la connaissance affranchit de l’obscurantisme ambiant. Maintenant les humains ont le choix entre d’une part la version créationniste de leur être qui stipule le lien entre la couleur de la peau et la malédiction noachique, et d’autre part la version scientifique basée sur les documents paléontologiques consultables de l’histoire de l’humanité. Si le monde fut créé par un dieu il y a 6011 ans, des hommes à peau noire construisaient des monuments défiant les connaissances actuelles avant cette période. De quoi confondre les idéologues suprématistes. Tels sont les faits têtus qu’aucune science digne de ce nom ne peut défaire à l’heure actuelle comme dirait Cheikh Anta Diop.
Répondre
Vous devez vous identifier pour commenter.